Genéricos y bioequivalencia…una elección acertada
Por: Julio César Alcubilla B.-
Un medicamento genérico define en el común denominador de la población, aquella versión de un fármaco que nos resulta más económica de los de marcas muy conocidas y muy utilizadas. Usualmente lo comprendemos "como copias más económicas"; que el farmaceuta luego de insistirle nos recomienda, porque reconoce en los mismos algunas enzimas del producto original. Sin embargo no está él ni nosotros capacitados, para saber hasta qué punto nos resultarán apropiados y cuáles estudios los avalan.
El término genérico en realidad ha de emplearse para denominar al medicamento cuyo ingrediente activo, sustancia o enzima es químicamente equivalente a sus contrapartes de marca en términos de ingredientes activos. Aunque difieran en presentación o excipientes, dichas drogas fueron definidas así por la FDA ((Food and Drug Administration), debido a la ausencia de diferencias significativas y de disponibilidad del ingrediente activo en el lugar de acción de la droga; determinándolas como bioequivalentes.
 El tema ha sido realmente polémico entre los diferentes organismos estatales a nivel mundial, realmente si logramos hacer una investigación por diferentes fuentes de información, llegaremos a algunas conclusiones, principalmente acerca de los efectos basados en parámetros clínicos relevantes, como la frecuencia cardiaca, presión arterial, medidas de laboratorio, utilización de los sistemas de salud o mortalidad. Principalmente cuando el índice terapéutico de esos medicamentos es estrecho.
El tema ha sido realmente polémico entre los diferentes organismos estatales a nivel mundial, realmente si logramos hacer una investigación por diferentes fuentes de información, llegaremos a algunas conclusiones, principalmente acerca de los efectos basados en parámetros clínicos relevantes, como la frecuencia cardiaca, presión arterial, medidas de laboratorio, utilización de los sistemas de salud o mortalidad. Principalmente cuando el índice terapéutico de esos medicamentos es estrecho.
La aprobación de un fármaco genérico por parte del organismo oficial competente, ha de ser basada en la evidencia científica, de que éste produce un efecto sobre los seres humanos esencialmente idéntico al del producto original. Muchos estudios de elevadas inversiones, son llevados a cabo para probar los fármacos genéricos nuevos y lograr determinar a través de los mismos, que contengan las cantidades adecuadas de principios activos y que están siendo fabricados según las normas de fabricación oficiales. Un aspecto importante es su liberación en el organismo a la misma velocidad y con el mismo alcance que los fármacos originales, con nombre comercial.
Ahora bien ¿todos los fármacos genéricos que consumimos o nos vemos irremediablemente motivados a adquirir, fundamentalmente por aquello del ahorro, son confiables, les han hecho tales estudios?; desafortunadamente no, no todos los medicamentos genéricos son tan satisfactorios como los de nombre comercial. Esto se debe en gran parte a que tales estudios no son realizados por todos los laboratorios, por otro lado al ser llevados a cabo, no están presentes en los empaques de los mismos y en el común de los casos, su biodisponibilidad no está muy clara. La biodisponibilidad es una prueba que identifica la fracción del medicamento administrado, que llega en forma activa a la circulación sistémica. Ésta suele variar de fabricante a fabricante y a veces entre lotes diferentes de un compuesto producido por el mismo fabricante. Por ello a la hora de elegir un genérico, debemos identificar, preguntar o indagar si posee estudios de bioequivalencia y que ocurre con su biodisponibilidad.
Lo realmente ético, es que cuando un laboratorio decide desarrollar un genérico, sus expertos en formulación deben diseñar éste producto utilizando los mismos principios activos a los del fármaco original, aunque probablemente usen componentes inactivos diferentes. Estos componentes inactivos, son añadidos generalmente para aumentar el volumen, logrando de ésta manera que la forma sólida sea lo suficientemente grande para manipularlo, evitando que dicha forma se disgregue durante el período de fabricación en lo referente a su uso, para contribuir a su disolución en el estómago o el intestino, o para que tenga sabor y color aceptables.
 ¿Qué ocurre con la prescripción de éste tipo de medicamentos?
¿Qué ocurre con la prescripción de éste tipo de medicamentos?
Debemos estar consientes como consumidores y pacientes, que no siempre el farmaceuta o especialista de la salud, nos está haciendo la mejor recomendación en éste sentido, y mucho menos "nuestro vecino". Los profesionales que prescriben deberían disponer de documentos oficiales sobre la equivalencia terapéutica entre fármacos genéricos y los presentados con nombre comercial, en Estados Unidos, la FDA (Food and Drug Administration), publica todos los años un libro sobre productos farmacéuticos aprobados con evaluaciones de equivalencia terapéutica. Esta sería una de las formas más rápidas de obtener información, acerca de la prescripción.
¿…Y qué ocurre con la patente de un fármaco en pro del paciente? En casi todos los países si una compañía desarrolla un nuevo fármaco se le concede una patente sólo para el propio fármaco, su proceso de fabricación o su utilización. Pero es común que el fabricante o laboratorio, posea más de una patente por fármaco, y puede por igual poseer además una patente del sistema que distribuye y libera el fármaco en la sangre.
Las patentes garantizan a la compañía los derechos exclusivos del fármaco durante un número determinado de años, alrededor de 10 años, desde el momento del descubrimiento al de su aprobación para uso en seres humanos. Esto obliga al mercado del fabricante, a que sólo se pueda comercializar el nuevo fármaco de manera exclusiva durante aproximadamente 7 años. A excepción de los fármacos contra el HIV/ SIDA y otros para tratar enfermedades con riesgo de muerte, los cuáles reciben con frecuencia una aprobación más rápida. Si la patente expira, otras compañías pueden vender una versión genérica del fármaco, con frecuencia a un precio mucho menor que el de la marca original. Allí radica el principal problema…
Los nuevos fármacos requieren estudios más extensos, más complejos y mucho más caros para demostrar que son seguros y efectivos. Además de ello, los laboratorios, al realizar tales estudios en el comprimido o la cápsula utilizada durante los ensayos clínicos y las fases del desarrollo del producto deben modificarse por razones comerciales. Las normas son diferentes para los fármacos de liberación regulada (de acción prolongada o de liberación sostenida). Dado que estos fármacos están sujetos a muchas más variaciones que los comprimidos y las cápsulas normales, los inspectores correspondientes necesitan extensas pruebas como requisito para la solicitud del nuevo fármaco, con el fin de que la compañía pueda comercializar una versión de acción prolongada de cualquier fármaco. Este trámite indispensable se aplica igualmente, aunque otra versión del fármaco con difusión regulada ya esté en el mercado. Este requisito ha retrasado la disponibilidad de versiones genéricas de algunos fármacos de difusión regulada. Sin embargo, la investigación se realiza en beneficio del consumidor.
¿Podrán éstos nuevos genéricos salir al mercado, con los estudios pertinentes?
Bioequivalencia y Biodisponibilidad en Venezuela La importancia de estos estudios, no escapa a los organismos competentes en nuestro país. Desde 2005 se dio inicio a las operaciones del Laboratorio de Bioequivalencia y Biodisponibilidad del Departamento de Química Medicinal del Instituto Venezolano de Investigaciones Científicas IVIC, el cual prestó su primer estudio a una empresa farmacéutica nacional
La importancia de estos estudios, no escapa a los organismos competentes en nuestro país. Desde 2005 se dio inicio a las operaciones del Laboratorio de Bioequivalencia y Biodisponibilidad del Departamento de Química Medicinal del Instituto Venezolano de Investigaciones Científicas IVIC, el cual prestó su primer estudio a una empresa farmacéutica nacional
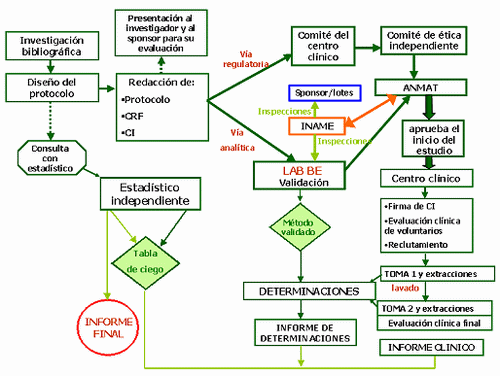
Este laboratorio realiza una evaluación de factibilidad del estudio acerca de un medicamento en específico, el cual consiste en determinar si los análisis requeridos, pueden realizarse en el laboratorio del instituto. Cumplida esta etapa se pasa a la elaboración de un informe denominado protocolo de bioequivalencia, en el que se evalúa y describe de manera minuciosa los aspectos técnicos del estudio, tomando en cuenta 3 etapas: una fase clínica, donde se trabaja con voluntarios sanos; una parte analítica, que se lleva a cabo en el laboratorio; y una parte estadística, donde los resultados son graficados y presentados al ente evaluador; que en nuestro país suele ser el Instituto Nacional de Higiene, organismo relacionado al Ministerio de Salud.
A nivel internacional, estos trabajos pueden tomar entre 6 y 8 meses. La elaboración de este protocolo ha servido para que los científicos del IVIC estimen el tiempo que tomará un estudio de bioequivalencia en nuestro país. El avance obtenido con el funcionamiento del Laboratorio de Bioequivalencia y Biodisponibilidad del IVIC coloca a Venezuela a la altura de países como Brasil, Argentina, México y Cuba, que han sido adoptados como patrones de referencia en este tipo de estudios, y de los que se ha recibido total apoyo.
Los estudios de bioequivalencia han alcanzado, en los últimos 15 años, una gran relevancia y, de hecho, Venezuela cuenta con la "Norma de Bioequivalencia y Biodisponibilidad", decretada en Gaceta Oficial Nº 38.499 del 14 de agosto de 2006.Un punto importante que se destaca de los estudios de bioequivalencia y biodisponibilidad es que se trabaja con substancias que posiblemente se han consumido con anterioridad, por eso se habla de voluntarios sanos y no de pacientes.
Con estos análisis se comprueba si existe "intercambiabilidad" del medicamento genérico, es decir, si su efecto terapéutico es exactamente igual al de marca, lo que supone que si un consumidor lo solicita en cualquier farmacia tendrá la seguridad de que con ambas opciones obtendrá el mismo efecto y será su decisión comprar el que desee, con la confianza puesta en el estudio realizado por el IVIC.
 Se pronuncian los especialistas… Dr. Carlos Capriles, médico internista, representante de Laboratorios Sandoz.-
Se pronuncian los especialistas… Dr. Carlos Capriles, médico internista, representante de Laboratorios Sandoz.-
¿Dr. Capriles hasta qué punto es aceptado por el cuerpo médico nacional, clínicas, hospitales y centros de salud, la farmacéutica genérica en un paciente sometido a tratamiento?
"El medicamento genérico, viene a resolver un problema de altos costos en la salud, por cuanto a los medicamentos originales, son productos de una investigación larga y muy costosa. El genérico atiende al 50% de la población que no tiene con que cubrir, totalmente los tratamientos. El genérico contiene el mismo principio activo, en la misma cantidad que el medicamento innovador, sin embargo el problema que vivimos los médicos constantemente es saber si podemos intercambiar, un medicamento innovador por un genérico."
"Esto se responde a través de una metodología, la cual es aceptada por todos los países desarrollados, la cual es el estudio de bioequivalencia. Medir la bioequivalencia equivale a comparar el comportamiento del medicamento genérico en el cuerpo del paciente, para poder hacer el intercambio. No todos los medicamentos genéricos tienen que tener estudios de bioequivalencia, no todos los medicamentos innovadores se pueden sustituir, pero en general la mayoría de los medicamentos, al tener bioequivalencia, podrían sustituirse y producir ahorros del 50% en adelante. Esto garantizaría que el paciente pueda completar su tratamiento y a nivel institucional, que el estado pueda costear esta inversión y dar acceso a todos en los centros de salud.".
Los estudios de bioequivalencia Dr. Capriles determinan a su vez la biodisponibilidad de un fármaco, sin embargo ¿cómo un paciente común podría reconocer que el genérico que va adquirir, cumple su promesa acoplada a la OMS, en cuanto a los estudios de concentración plasmática y la biodiversidad?
"Probablemente estemos tocando términos bastantes complejos ante la opinión pública y el paciente promedio, más considerando la escasa información acerca del tema. La bioequivalencia lo que compara es si hay o no una diferencia entre la cantidad de medicamento que llega a la sangre y un grupo de personas que se someten a esa prueba. No son pacientes, son sujetos sanos comparando el genérico con el innovador. La biodisponibilidad de niveles en sangre, se lleva a cabo en un laboratorio certificado, a través de un análisis estadístico muy riguroso y si no hay diferencia se dicen que son bioequivalentes".

"Ahora bien, esta decisión no ha de tomarla el paciente sino el médico, obviamente los médicos debemos estar más tranquilos si cuando estamos recetando sabemos que el medicamento tiene bioequivalencia. Nos toca a los médicos, solicitarles a los laboratorios, los estudios de bioequivalencia. Por ello estamos llevando a cabo un tour de bioequivalencia, donde estamos realizando, actualización de los colegas y los profesionales de salud".
Obviamente, deberíamos en Venezuela y en el resto de países de Latinoamérica, consultar a nuestro médico o especialista, sin embargo más nos guiamos por la recomendación de un vecino o familiar, nos automedicamos; mas si se trata de una enfermedad o patología común…¿Cómo identificar un falso genérico, cómo hacerlo con un medicamento con licencia , cómo identificar a un medicamento original y finalmente como asimilar el precio y la mejor relación Inversión/confianza/beneficio?
"En primer lugar, en Venezuela no debemos preocuparnos por falsos genéricos o genéricos no confiables, debido a que legalmente no existe. Los medicamentos llamados innovadores provienen de una investigación original que lleva más o menos de 8 a 10 años hacerla, estos son llamados comúnmente originales. Esto no implica que existan medicamentos originales y medicamentos falsos, los medicamentos innovadores provienen de una investigación original y cuesta aproximadamente US$ 600 a 800 millones. Estos son los primeros que vemos y salen con su marca al mercado, por ello son productos de un costo alto, porque se debe cubrir todo el proceso de investigación y preparar la próxima investigación. A estos medicamentos se les concede una patente, la cual dura entre 15 y 20 años, repartidos entre los 10 primeros años invertidos en investigación y luego alrededor de 10 años, para comercializarlo, en el que el medicamento tiene el monopolio de su clase, permitiéndoles a los laboratorios recuperar el dinero que invirtieron".
 "El término licencia, tiene que ver que cuando un medicamento tiene patente, la patente es cedida a otro. La licencia tiene que ver más con la propiedad intelectual y no con la investigación. Ahora profundicemos en los medicamentos que llamamos, originales y copias, en Venezuela la autoridad regulatoria permite que una vez que un medicamento innovador, introdujo toda su data, vengan las segundas marcas y los genéricos. Estos son los que masifican el componente activo, o que descubrió el medicamento innovador. Esto permite poner a la venta medicamentos de un costo menor, para que todo el mundo tenga acceso a ellos, no queriendo decir que sean de mala calidad. En Venezuela, la autoridad es muy estricta a la hora de registrar un medicamento. Todos los medicamentos en nuestro país, deben tener la prueba de disolución, que es la prueba que garantiza que la tableta se va a disolver. Finalmente tenemos un grupo muy pequeño de medicamentos innovadores, que su margen es tan estrecho, que a veces con el estudio de bioequivalencia, no es suficiente para realizar el intercambio. Estos son medicamentos no sustituibles".
"El término licencia, tiene que ver que cuando un medicamento tiene patente, la patente es cedida a otro. La licencia tiene que ver más con la propiedad intelectual y no con la investigación. Ahora profundicemos en los medicamentos que llamamos, originales y copias, en Venezuela la autoridad regulatoria permite que una vez que un medicamento innovador, introdujo toda su data, vengan las segundas marcas y los genéricos. Estos son los que masifican el componente activo, o que descubrió el medicamento innovador. Esto permite poner a la venta medicamentos de un costo menor, para que todo el mundo tenga acceso a ellos, no queriendo decir que sean de mala calidad. En Venezuela, la autoridad es muy estricta a la hora de registrar un medicamento. Todos los medicamentos en nuestro país, deben tener la prueba de disolución, que es la prueba que garantiza que la tableta se va a disolver. Finalmente tenemos un grupo muy pequeño de medicamentos innovadores, que su margen es tan estrecho, que a veces con el estudio de bioequivalencia, no es suficiente para realizar el intercambio. Estos son medicamentos no sustituibles".
¿Cómo se lleva a cabo un estudio de Bioequivalencia en Venezuela?
"Este es el método aceptado por las agencias regulatorias de todos los países, para evaluar la calidad de un medicamento. Constituye en comparar, el comportamiento del principio activo en las dos formas farmacéuticas. Es decir cómo se comporta el ingrediente activo que realmente hace el trabajo terapéutico, en la presentación innovadora y la presentación de prueba, que en éste caso sería el genérico o el similar. Se seleccionan al azar un grupo de sujetos sanos, y en iguales condiciones se le administra a cada grupo, el medicamento innovador o el medicamento de prueba, y se mide en sangre por cerca de 18 mediciones en sangre. Identificando como se absorbe la droga y que comportamiento tiene en el organismo. Esto se lleva a cabo bajo unas condiciones muy estrictas y rigurosas, luego se someten a un análisis estadístico para garantizar que la ausencia de diferencia no es producto del azar, y una vez que está demostrado, se puede decir entonces que el comportamiento, de éste genérico en comparación con el innovador, en estos sujetos sanos fue similar. Un estudio de bioequivalencia bien hecho, permite la intercambiabilidad del medicamento, y esto es aceptado por los entes de regulación locales y la OMS (Organización Mundial de la Salud)."
¿Dr. Capriles, no puede considerarse una utopía que el medicamento genérico considerado bioequivalente, debió comprobar su equivalencia terapéutica con la formulación en referencia?
"Veámoslo de ésta manera, en los años 70, empezaron a surgir medicamentos con el mismo principio activo del innovador. Llegaron a tener fallas, esto pasó con un medicamento para el corazón llamado Digoxina. Se comenzó a medir la presencia de Digoxina en la sangre, y se comprobó, que en esa época de los 70, había una diferencia entre dos medicamentos, que aparentemente, se veían iguales. Las autoridades regulatorias de ese país, paso este caso a la conciencia y autoridades regulatorias mundiales, quienes hallaron la manera científica de demostrar y de estudiar, el comportamiento de dos formulaciones, que contienen el mismo principio activo, llegando a los estudios de bioequivalencia".
Rosangela de Sarios, Gerente de Asuntos Registrables de Laboratorios Sandoz.
¿Determina la bioequivalencia que un genérico sea adquirido en el mercado por ser para la población 100% confiable?
"Los estudios de Bioequivalencia son estudios clínicos, que simplemente buscan la comparación de dos formulaciones. Las sustancias activas, seguirán siendo las mismas. En los países de primer mundo, los medicamentos genéricos salen al mercado, una vez que hayan expirado los medicamentos con patentes. Y estos pueden entrar al mercado a través de los estudios de bioequivalencia, esto ayuda a dar confiabilidad en el medicamento que se está adquiriendo".
Dentro de los estudios de bioequivalencia, se utiliza el método trapezoidal, el cual determina la concentración plasmática del medicamento, desde el momento en que fue dosificado hasta el último tiempo de muestreo. Esto permite determinar, que el fármaco que ingresó al organismo, como fue absorbido.
 ¿Los genéricos de Sandoz y sus estudios de Bioequivalencia, contemplan éste método?
¿Los genéricos de Sandoz y sus estudios de Bioequivalencia, contemplan éste método?
"Efectivamente existe una normativa internacional de bioequivalencia, e incluso una normativa local, que en Venezuela se publicó en el 2006, que establece éste método trapezoidal, para determinación del área bajo la curva, no realmente para la concentración plasmática. La concentración plasmática es un parámetro que se mide en inversión al tiempo; tomando en cuenta cual es la concentración de la droga en sangre en un tiempo determinado, se va estructurando una curva que posteriormente a través de éste método trapezoidal, se calcula el área bajo la curva. Determinando una media, con respecto a la eliminación de la droga, que no siempre llega a conclusiones exactas. Estos métodos clínicos son muy confiables, Sandoz lo utiliza".
En consecuencia estas pruebas son responsabilidad del laboratorio que registra el producto, dentro de este contexto ¿por qué el genérico de Sandoz, ha de ser la elección confiable por parte del paciente?
"Existe una normativa farmacológica, que exige en algunos casos estudios de farmacocinética para productos innovadores, para los genéricos existe una norma ya explicada, que no obstante no se aplica en su totalidad. Se está implementando paulatinamente, todavía no existe toda la infraestructura en el Estado, para solicitarles a todos los productos, que presenten éstos estudios de bioequivalencia. En éste sentido existe una lista de medicamentos que se consideran, bien sea por su estrecho margen terapéutico, o los que su cantidad de la droga es muy estrecha, se les está solicitando, tales estudios. Laboratorios Sandoz desde el año 2000, está presentando regularmente todos los estudios de bioequivalencia en todos nuestros productos, debido a esto tenemos un plazo adelante".
Fuente: Julio César Alcubilla B.-www.saludytecnologia.net