Progeria
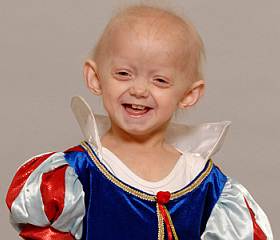
La progeria es una enfermedad genética extremadamente rara, caracterizada por un envejecimiento brusco y prematuro, cuya forma más común ataca a niños entre su primer y segundo año de vida.
Se estima que afecta a uno de cada 8 millones de recién nacidos vivos. No se ha evidenciado preferencia por ningún sexo en particular, pero se han comunicado muchos más pacientes de tipo étnico caucásico (97% de los pacientes afectados).
La progeria puede afectar diferentes órganos y tejidos: hueso, músculos, piel, tejido subcutáneo y vasos.
La forma más severa de esta enfermedad es el llamada síndrome de Hutchinson-Gilford, por los dos científicos que primero la analizaron: Jonathan Hutchinson, el primero en describirla en 1886, y Hastings-Gilford, quien realizó diferentes estudios acerca de su desarrollo y características en 1904.
Descripción detallada
Progeria:
• Del griego pro, "hacia, a favor de" y geron, "viejo".
• Sindrome por Envejecimiento Prematuro.
• Causa Genética (Monogénica).
• Incidencia
– Uno en 8 millones de RN (EUA 1915-1967).
– Uno en 4 millones de RN (Holanda 1900-2005).
• Se han reportado pacientes en casi todos los países, de todos los continentes y en todos los grupos étnicos.
• Tres principales formas clínicas:
a- P. de la Infancia o Clásica
b- P. Congénita o Neonatal
c- P. del Adulto
(A) Progeria de la infancia o progeria clásica.
• La forma más frecuente y severa.
• Apariencia física normal al nacimiento.
• Bajo peso para edad gestacional: 2500 gr. o menos.
• Talla normal para edad gestacional.
• Edad de comienzo : 1 – 3 años.
Características clínicas
• En el 1er. año:
– Retardo severo y posteriormente,detención del crecimiento.
– Desarrollo normal: No hay retardo psíquico ni motor.
– Desproporción cráneo – cara, por Macrocefalia; "abombamiento" o protuberancias frontales laterales.
– Venas craneales prominentes.
– Cierre tardío de la Fontanela Ant.
• 1er. al 3er. año (facies característica):
– Triangular, de apariencia envejecida.
– Puente nasal angosto y alto.
– Nariz estrecha y puntiaguda.
– Labios delgados, boca pequeña.
– Micrognatia, retrognatia.
– Cejas y pestañas escasas o ausentes.
– Ojos prominentes.
– Pabellones auriculares displásicos.
Otros signos:
– Alopecia parcial, luego total.
– Erupción y caída tardía de dientes primarios.
• Después:
– Piel seca, pigmentada; disminución del tejido celular subcutáneo (adiposo).
– Esclerodermia en región inferior del abdomen y proximal de muslos.
– Caracteres sexuales secundarios poco desarrollados.
– Clavículas cortas y distróficas.
– Osteolisis de las falanges distales.
– Uñas cortas y distróficas en manos y pies.
• Más tardías:
– Hipoacusia conductiva para frecuencias bajas.
– Erupción parcial de dientes secundarios; malposición dentaria.
– Articulaciones prominentes (engrosadas), con contracturas y rigidez. Osteoartritis.
– Osteopenia. Osteoporosis.
– Coxa valga.
– Marcha inestable, con aumento de la base de sustentación.
– Voz de tono agudo.
Manifestaciones cardiovasculares tempranas:
– Hipertensión arterial.
– Cardiomegalia.
– Ateroesclerosis / Ateromatosis severa.
Complicaciones cardiovasculares:
– Infarto del miocardio.
– Isquemia cerebral tranisitoria, accidente cerebrovascular.
– Insuficiencia cardíaca.
Pronóstico:
Fallecen, en promedio, hacia los 13 años de edad.
Rango de edad= 6 – 20 años.
Diagnóstico
Se basa fundamentalmente en el examen físico y el reconocimiento de las características clínicas propias de la progeria clásica o infantil.
Cuando sea factible, puede solicitarse el análisis y secuenciación del gen mutante.
Genética
Gen LMNA (locus: 1q22). Único gen con la mutación causante de la Progreria Clásica o Infantil.
– La mutación c.1824C-T es una mutación de punto (sustitución del nucleótido C por T en esa posición) y se encuentra en casi 100% de los pacientes con Progeria Clásica.
– Las progerias atípicas son causadas por otras mutaciones en el gen LMNA.
– El gen normal codifica la proteína Laminina (Lámina A/C), principal componente de la membrana nuclear, ubicada en su cara interna.
Etipatogenia
La mutación c.1824C-T codifica una proteína anormal llamada Progerina, que tiene 50 amino-ácidos menos que la Laminina. La Progerina produce la disrupción de la Lámina A/C, alteración de la membrana nuclear y de su función.
Los cultivos de fibroblastos de pacientes con Progeria Clásica muestran inicialmente crecimiento celular acelerado, seguido de disminución rápida en la proliferación. Hay disminución del tamaño de las células, con membranas celulares gruesas, pérdida del contacto célula a célula, con inhibición del crecimiento y agrupamiento de las mismas. Los núcleos muestran múltiples anormalidades estructurales como herniaciones y lóbulos, y defectos en la organización de la heterocromatina.
Estos trastornos nucleares producen efectos profundos en la integridad del tejido conectivo, componente esencial de varios órganos y sistemas que presentan las características clínicas del síndrome.
Asesoramiento genético
– Frecuentemente los pacientes con Progeria Clásica son casos esporádicos o únicos, productos Heterocigotos por Neo-mutación c.1824C-T) Autosómica Dominante.
– Los padres no están afectados y su riesgo de recurrencia es bajo. Se estima en 1:500 (0,2%), considerando la probabilidad del mosaicismo germinal en uno de los progenitores.
– La Neo-mutación está asociada a edad paterna avanzada.
– Los enfermos no viven lo suficiente para tener descendencia.
Son muy escasos los pacientes descritos con sobrevida por encima de los 13 – 20 años de edad. Ejemplo excepcional es un hombre con Progeria Clásica, diagnosticado en la infancia, que falleció a los 47 años, por Infarto del Miocardio. Tenía un hijo con el mismo sindrome y otro sano. Esto corresponde a un trastorno Autosómico Dominante, en donde el padre enfermo es Heterocigoto y su riesgo de recurrencia es 1 : 2 (50%) en cada concepción. El gen mutante tiene penetrancia completa.
El Asesoramiento Genético Integral se extiende más allá de informar el riesgo de recurrencia a los padres de un niño enfermo. También debemos tratar aspectos como:
– Planificación Familiar Temporal.
– Esterilización quirúrgica voluntaria.
– Evaluaciones periódicas por varios especialistas.
– Apoyo emocional al paciente y sus padres, y, si es necesario, consulta con psicólogo ó psiquiatra.
– Aclarar dudas y errores de interpretación.
Seguimiento, manejo y tratamiento del paciente
Hechas las evaluaciones iniciales y de control, se aplicarán los tratamientos conocidos para disminuir o aliviar las manifestaciones clínicas relevantes y las complicaciones que puedan presentarse.
Terapias en investigación
Algunas en etapa de Pruebas Clínicas:
– Lonafamib, un antineoplásico que actúa como inhibidor de la farnesiltransferasa (FTI). Eficacia probada en ratones modelos.
– Prevastatina (estatina inhibidora de la HMG-CoA reductasa), en combinación con el Zoledionate, que inhibe la síntesis del farnesil-pirofosfato.
– Inducción de células madres o células pluripotenciales (stem cells) a partir de fibroblastos obtenidos de pacientes con Progeria Clásica han revertido las alteraciones nucleares y mostrado ausencia de la Progerina propias del sindrome.
– Muy nuevos tratamientos relacionados con la rapamicina en el National Human Genome Research Institute, los cuales podría proporcionar un beneficio clínico para los niños con HGPS
Una vez resueltos algunos problemas, las las prueban clínicas deberán dar resultados positivos en los enfermos.
Perspectivas
Como la Progerina también se acumula en el envejecimiento de personas sin progeria, es de esperar que los tratamientos que se apliquen para la Progeria sirvan también en parte para prolongar los años de vida útil del adulto, deteniendo el envejecimiento fisiológico, especialmente el vascular.
Otras formas de progeria
B• Progeria congénita o neonatal
– Signos clínicos reconocibles al nacimiento o pocodespués, similares a los de la Progeria Clásica.
– Los pacientes fallecen pronto, entre 3 – 5 años.
– Este sindrome es Autosómico Recesivo.
C• Progeria del adulto
– Los signos clínicos característicos se presentan entre los 15 y 30 años de edad.
– Es un sindrome Autosómico Recesivo.
Foto de cabecera: ZUMA Press.
Artículos relacionados:
– Medicina regenerativa: avances en células madres contra la progeria.
– Las 10 anomalías más raras en medicina.
– The Curious Case of Benjamin Button.
– Esperanza contra la progeria y para ralentizar el envejecimiento.
Escuche una completa disertación sobre la progeria (síndrome de envejecimiento prematuro Hutchinson-Gilford) y las esperanzas que ofrecen recientes investigaciones para quienes sufren esta condición, con el Dr. Frank G. Hammond, Médico Genetista, eminente investigador y académico de Genética Médica, quien detalla las características de la progeria, que la produce y su incidencia, además, asoma importantes desarrollos de la investigación médica que podrían ralentizar el deterioro característico del envejecimiento prematuro; en entrevista conducida por Peter T; directamente aquí o en su plataforma de podcast preferida
• ApplePodcasts: http://j.mp/AppleProgeria
• RadioPublic: http://j.mp/RadioPublicProgeria
• Spotify: http://bit.ly/SpotifyProgeria
• Whooshka: http://j.mp/WhooshkaProgeria
• Stitcher: http://j.mp/StitcherProgeria
• También estamos en GooglePlayMusic (http://bit.ly/THPGooglePlay) y Pocketcast (https://pca.st/fP2H).
Fuente: Dr. Frank Hammond y wikipedia.es