La FCC toma partido por un internet abierto y neutral

… O como las batallas se tienen que dar:
Nadie te callará, nadie te censurará, nadie te quitará tu libertad, pero depende de ti dar la batalla por tu libertad, nadie peleará por ti (Dichos Perdidos de Muadib)
El día de hoy la Federal Communications Commission (@FCC) ha dado un paso claro por el #internetlibre la necesidad de contar con un #internetneutra. Una votación cerrada (3 a 2), pero una votación ganada. Ciertamente que las telecom deberán apelar (y deberán porque ya dijeron que lo harían), pero esta batalla se ganó.
Tom Wheeler, Chairman de la FCC dijo "No one, whether government or corporate, should control the open and free access to the Internet." En buen cristiano: "Nadie, ni gobierno ni corporaciones, deben controlar el libre y abierto acceso a Internet". Es pués una declaración sencilla, simple, directa y, sobre todo, con una fuerza clara de qué es lo que implica el internet para la democracia y la necesidad que sea neutra como elemento básico de una #internetlibre.
Pero no solo se quedaron en la declaración retórica avanzaron a declarar el Internet como un servicio público, bajo el Título II. El desarrollo de la industria de contenidos como elemento económico y el ejercicio pleno de la libertad de expresión como instrumento democrático y de ejercicio de #DDHH es lo que están detrás de esta decisión.
¿Qué dijo la FCC?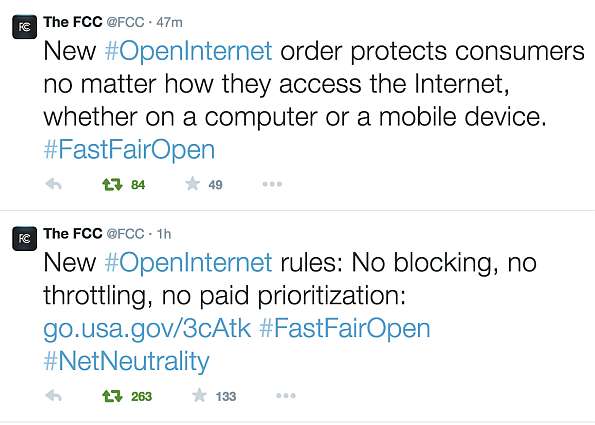
¿Qué dijo la Casa Blanca?
Menudo problema que tendrán ciertamente que lidiar para equilibrar este avance con todas acusaciones que tiene la NSA y el sistema de inteligencia norteamericano de acceder a comunicaciones y patrones de uso de internet. Discusión que se debe dar también. No solo un #internetabierto sino un #internetseguro y sin violaciones de #DDHH por parte de ningún gobierno ni corporación.
Hoy es un día para alegrarnos la #neutralidaddelared ganó esta batalla. Es un día para descansar, pero esta guerra recién empieza.
Mas Información:
– FCC votes to protect the internet with Title II regulation
– La FCC da el paso de gigante definitivo hacia la neutralidad de la red
– ISOC – Thoughts from @SAWentworth on today's FCC Net Neutrality Ruling.
Encuentre más del mismo autor en lexdigitalis.lamula.pe y en la cuenta Twitter @coyotegris.
Fuente: Erick I.A. / @coyotegris / lexdigitalis.lamula.pe